Input interpretation
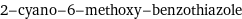
2-cyano-6-methoxy-benzothiazole
Chemical names and formulas
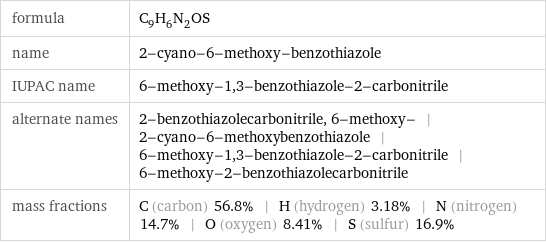
formula | C_9H_6N_2OS name | 2-cyano-6-methoxy-benzothiazole IUPAC name | 6-methoxy-1, 3-benzothiazole-2-carbonitrile alternate names | 2-benzothiazolecarbonitrile, 6-methoxy- | 2-cyano-6-methoxybenzothiazole | 6-methoxy-1, 3-benzothiazole-2-carbonitrile | 6-methoxy-2-benzothiazolecarbonitrile mass fractions | C (carbon) 56.8% | H (hydrogen) 3.18% | N (nitrogen) 14.7% | O (oxygen) 8.41% | S (sulfur) 16.9%
Lewis structure
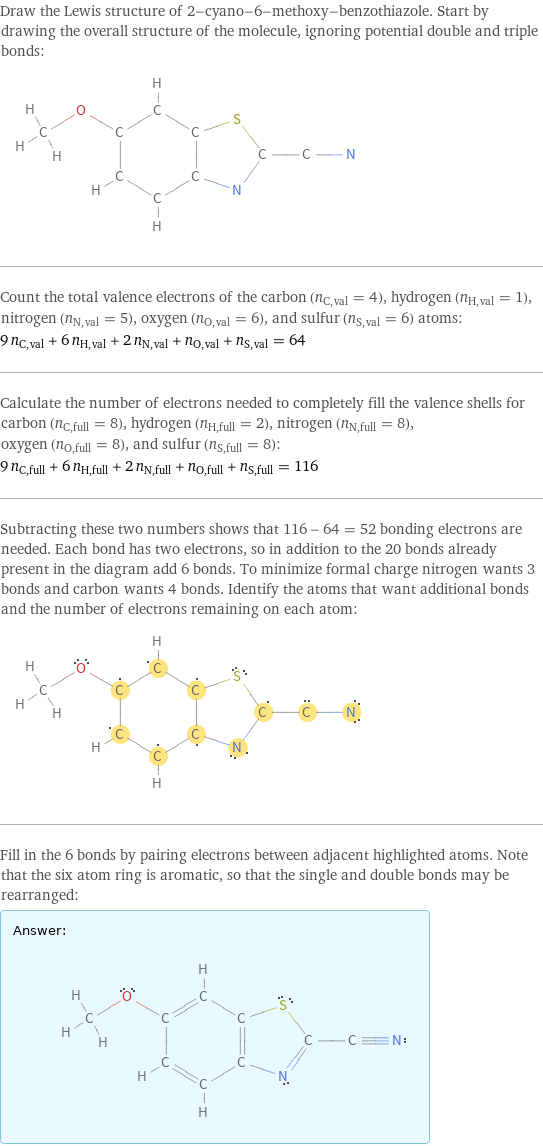
Draw the Lewis structure of 2-cyano-6-methoxy-benzothiazole. Start by drawing the overall structure of the molecule, ignoring potential double and triple bonds: Count the total valence electrons of the carbon (n_C, val = 4), hydrogen (n_H, val = 1), nitrogen (n_N, val = 5), oxygen (n_O, val = 6), and sulfur (n_S, val = 6) atoms: 9 n_C, val + 6 n_H, val + 2 n_N, val + n_O, val + n_S, val = 64 Calculate the number of electrons needed to completely fill the valence shells for carbon (n_C, full = 8), hydrogen (n_H, full = 2), nitrogen (n_N, full = 8), oxygen (n_O, full = 8), and sulfur (n_S, full = 8): 9 n_C, full + 6 n_H, full + 2 n_N, full + n_O, full + n_S, full = 116 Subtracting these two numbers shows that 116 - 64 = 52 bonding electrons are needed. Each bond has two electrons, so in addition to the 20 bonds already present in the diagram add 6 bonds. To minimize formal charge nitrogen wants 3 bonds and carbon wants 4 bonds. Identify the atoms that want additional bonds and the number of electrons remaining on each atom: Fill in the 6 bonds by pairing electrons between adjacent highlighted atoms. Note that the six atom ring is aromatic, so that the single and double bonds may be rearranged: Answer: | |
3D structure
3D structure
Basic properties
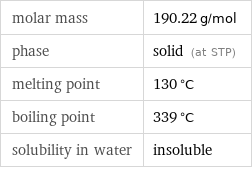
molar mass | 190.22 g/mol phase | solid (at STP) melting point | 130 °C boiling point | 339 °C solubility in water | insoluble
Units

Molecular properties

dipole moment | 5.777 D (debyes) total molecular energy | -925.6 E_h (Hartree energies) nuclear repulsion energy | 799.2 E_h (Hartree energies) HOMO energy | -0.3325 E_h (Hartree energies) LUMO energy | 0.05901 E_h (Hartree energies) HOMO-LUMO gap | 0.3916 E_h (Hartree energies) (computed using Hartree-Fock with a 6-31G(d) basis set)
Chemical identifiers

CAS number | 943-03-3 PubChem CID number | 342109 PubChem SID number | 24855838 SMILES identifier | COC1=CC2=C(C=C1)N=C(S2)C#N InChI identifier | InChI=1/C9H6N2OS/c1-12-6-2-3-7-8(4-6)13-9(5-10)11-7/h2-4H, 1H3 MDL number | MFCD00010537